Intervista ad Antonio Musarò, nuovo studio sull'origine della SLA
Individuato meccanismo molecolare alla base smantellamento giunzione neuromuscolare (NMJ) che si verifica in molte patologie tra cui la SLA.
Recentemente sulla rivista scientifica Antioxidant and redox signaling (Ars) è stato pubblicato lo studio “Muscle Expression of SOD1G93A Triggers the Dismantlement of Neuromuscular Junction via PKC-Theta” coordinato dal prof. Antonio Musarò dell'università Sapienza di Roma, con la collaborazione di Fondazione Roma, IIT-Sapienza, Istituto Pasteur-Italia e Telethon, che ha individuato il meccanismo molecolare responsabile dello smantellamento della giunzione neuromuscolare (NMJ) che si verifica in molte patologie e alterazioni patologiche tra cui la SLA (sclerosi laterale amiotrofica).
Abbiamo chiesto al prof. Musarò di spiegarci meglio sia la sua attività come docente universitario/ricercatore sia i risultati conseguiti con questo studio e le possibili implicazioni per la cura di determinate malattie.
Intervista al prof. Antonio Musarò - All'origine della SLA un "errore di comunicazione" tra muscolo e nervo
1) Antonio Musarò è professore di medicina e biotecnologie presso il Dipartimento di Scienze anatomiche, istologiche, medico-legali e dell’apparato locomotore della Sapienza. Ci spiega nel dettaglio qual è il suo lavoro e il suo settore di ricerca?
Il mio lavoro è un pò come quello di Ulisse raccontato da Dante Aligheri nella divina commedia: navigatore ed esploratore della conoscenza.
Il mio lavoro si svolge tra il laboratorio di ricerca e le aule universitarie dove insegno diversi moduli didattici. In particolare, insegno istologia ed embriologia agli studenti del corso di laurea magistrale in medicina e chirurgia e biotecnologie cellulari agli studenti del corso di laurea specialistica in biotecnologie mediche. Sono anche docente in diversi corsi di laurea triennale e nel dottorato di ricerca in Morfogenesi e ingegneria tissutale
2) Lei dirige un laboratorio di ricerca e coordina l’unità di ingegneria tissutale e il gruppo di ricerca neuromuscolare presso l’Università Sapienza, può spiegarci nel dettaglio in cosa consistono le attività di ricerca che vengono svolte nel laboratorio Musarò e quali sono i campi di applicazione?
Nel mio laboratorio ci occupiamo fondamentalmente di ricerca di base. Studiamo l'omeostasi e la rigenerazione muscolare in condizioni normali e patologiche. In particolare, la nostra ricerca si concentra su tre aree principali:
a) Studiare il ruolo del microambiente nella rigenerazione muscolare e capire come un microambiente ostile possa influenzare negativamente l'attività delle cellule staminali.
b) Studiare l'interazione fisiopatologica tra muscolo e nervo in condizioni normali e in diverse patologie degenerative.
c) Ingegneria tissutale. Nel mio laboratorio abbiamo recentemente sviluppato un costrutto tridimensionale di tessuto muscolare, chiamato X-MET che si sta mostrando un ottimo modello sperimentale per la ricerca di base e pre-clinica e una buona base di partenza per applicazioni in medicina rigenerativa
3) Recentemente sulla rivista Antioxidant and redox signaling (Ars) è stato pubblicato lo studio “Muscle Expression of SOD1G93A Triggers the Dismantlement of Neuromuscular Junction via PKC-Theta”, da lei coordinato, riguardante l’individuazione del meccanismo molecolare responsabile dello smantellamento della giunzione neuromuscolare (NMJ) e la dipendenza di tale smantellamento da una specifica proteina. Ci spiega di cosa tratta nel dettaglio lo studio che è stato pubblicato sulla rivista Ars e che cos’è e a cosa serve una giunzione neuromuscolare?
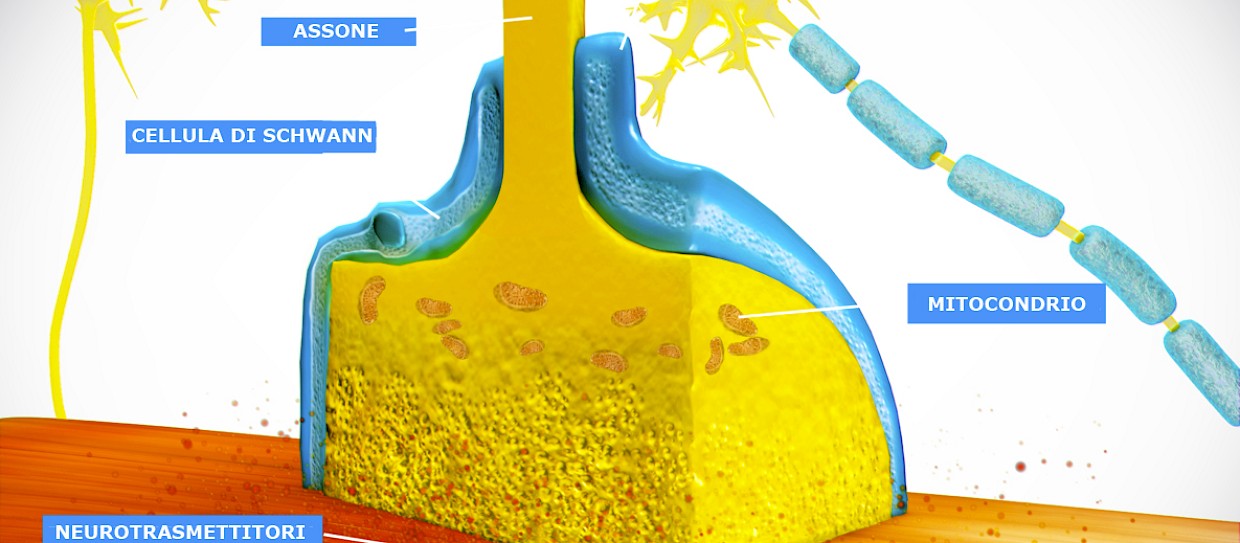
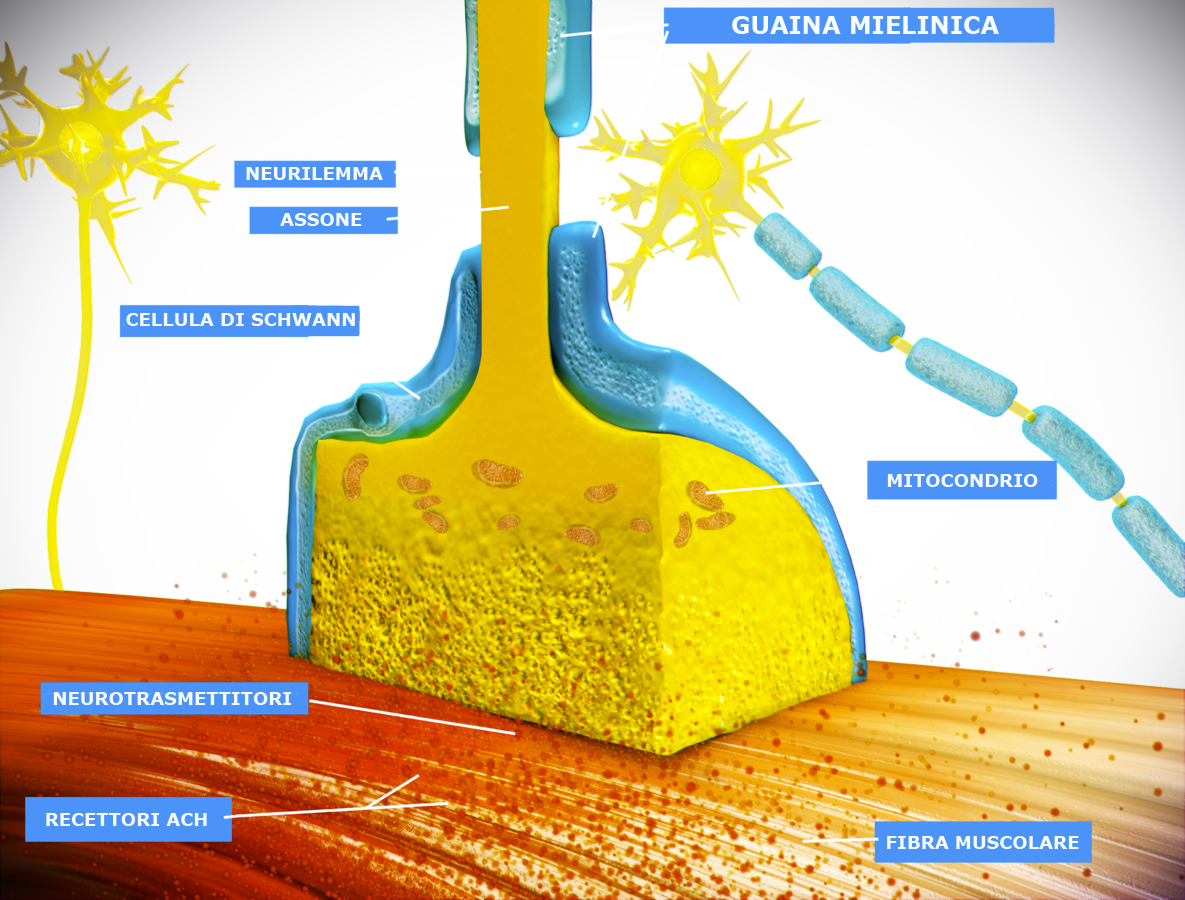 Immagine - Una giunzione neuromuscolare è una sinapsi chimica formata dal contatto tra un motoneurone e una fibra muscolare. Il motoneurone è in grado di trasmettere un segnale alla fibra muscolare grazie alla giunzione neuromuscolare causando in questo modo una contrazione muscolare. Immagine modificata. Original picture by Doctor Jana - http://docjana.com/#/nmj, CC BY 4.0, https://commons.wikimedia.org/w/index.php?curid=46835961
Immagine - Una giunzione neuromuscolare è una sinapsi chimica formata dal contatto tra un motoneurone e una fibra muscolare. Il motoneurone è in grado di trasmettere un segnale alla fibra muscolare grazie alla giunzione neuromuscolare causando in questo modo una contrazione muscolare. Immagine modificata. Original picture by Doctor Jana - http://docjana.com/#/nmj, CC BY 4.0, https://commons.wikimedia.org/w/index.php?curid=46835961
Le giunzioni neuromuscolari sono la regione di comunicazione tra muscolo e nervo e rappresentano un vero e proprio ponte funzionale tra i due tessuti. Infatti, ricevendo stimoli sia fisiologici che patologici dai due tessuti, cioè dal muscolo e dal nervo, consentono agli stessi di funzionare e comunicare in modo corretto.
Tuttavia, nel quadro degli studi finora condotti rimaneva irrisolto il problema se lo smantellamento delle NMJ, che si verifica in diverse condizioni patologiche (es. invecchiamento, SLA, distrofie muscolari), fosse un evento riconducibile direttamente a livello delle cellule nervose, quali i neuroni motori, oppure se possa verificarsi indipendentemente dalla loro degenerazione.
Il nostro studio ha tentato di rispondere a questa domanda e abbiamo realizzato un modello sperimentale ad hoc. L’obiettivo era quello di indagare se una alterazione che parte dal muscolo potesse compromettere il “mantenimento” della giunzione neuromuscolare e quindi la comunicazione muscolo-nervo. Abbiamo quindi individuato il meccanismo molecolare alla base dello smantellamento e scoperto che questo dipende dall’attivazione di una proteina chinasi, conosciuta come PKC theta. Conseguentemente, un suo “silenziamento” farmacologico ha permesso di preservare le giunzioni neuromuscolari e di promuovere un mantenimento della massa e forza muscolare nei topi trattati.
4) Prima della sua ricerca, svolta in collaborazione con la Fondazione Roma, IIT-Sapienza, Istituto Pasteur-Italia e Telethon, che ha portato all’individuazione del meccanismo molecolare alla base dello smantellamento della giunzione neuromuscolare, su cosa si basavano le ricerche precedenti riguardanti l’individuazione dei meccanismi patogenici caratterizzanti della sclerosi laterale amiotrofica (SLA)? Quali erano i problemi irrisolti in tali ricerche?
Fino a poco tempo fa la comunità scientifica concordava sul fatto che la SLA colpisse esclusivamente i motoneuroni e che i muscoli volontari fossero solo un bersaglio secondario della malattia. Recentemente, però, questo "dogma" ha cominciato a scricchiolare: è diventato sempre più chiaro infatti che il danno riguarda anche altri tessuti e cellule. La SLA può avere una componente genetica; circa il 10% dei pazienti è affetto da una forma ereditaria della malattia che, nel 20% dei casi, è dovuta a una forma mutata di una proteina, la superossido dismutasi 1 (Sod1). In condizioni normali Sod1 funziona come uno "spazzino" molecolare, eliminando dalle cellule i pericolosi radicali liberi.
Quando però la proteina Sod1 è mutata diventa tossica, favorendo la degenerazione cellulare. Le nostre ricerche hanno dimostrato per la prima volta che anche il muscolo scheletrico può essere tra i tessuti danneggiati direttamente dall'effetto tossico di Sod1 e che può quindi contribuire allo sviluppo della patologia. Abbiamo creato un modello animale della malattia in cui la versione "sbagliata" della proteina Sod1 veniva prodotta soltanto nel muscolo scheletrico e non nei motoneuroni. Abbiamo dimostrano che l’espressione localizzata del gene mutato Sod1 nei muscoli volontari dei topolini era in grado di indurre i segni pre-sintomatici della SLA, sia a livello muscolare (es. atrofia muscolare e significativa riduzione della forza muscolare), sia a livello delle giunzioni neuromuscolari e del midollo spinale, quali attivazione della microglia e di citochine infiammatorie, che normalmente precedono la degenerazione dei motoneuroni.
Da comprimario a coprotagonista quindi: il muscolo scheletrico si rivela tra i bersagli principali della proteina tossica responsabile di buona parte delle forme ereditarie di SLA.
5) Ci spiega quale è l’obiettivo che vi siete prefissi con gli esperimenti di laboratorio eseguiti su modelli murini, in che modo sono stati condotti tali esperimenti e quali sono state le criticità e i risultati che sono stati raggiunti?
L’obiettivo del nostro studio è quello di capire i meccanismi patogenetici di malattie degenerative come la SLA, la distrofia muscolare e la sarcopenia.
I modelli sperimentali usati da tanti laboratori di ricerca hanno permesso di capire sempre meglio i meccanismi molecolari e cellulari coinvolti in molte patologie ed in alcuni casi di arrivare anche a curare o rallentare la progressione di una malattia.
La nostra ricerca è partita da una considerazione e una constatazione: gli sforzi scientifici e sperimentali volti a “curare” la SLA partendo fondamentalmente dal motoneurone non hanno portato ai risultati attesi e ad oggi non esistono terapie risolutive, suggerendo che gli scarsi risultati ottenuti con la terapia convenzionale siano proprio dovuti ad una incompleta conoscenza delle basi molecolari e cellulari della malattia stessa. La constatazione era che molti dei difetti pre-sintomatici della SLA riguardano in primis i muscoli volontari e soltanto in una seconda fase si osserva la degenerazione dei neuroni motori, suggerendo che il muscolo scheletrico fosse uno dei bersagli primari della malattia e della tossicità della proteina Sod1 mutata, responsabile del 10% dei casi di SLA familiare.
E’ interessante inoltre notare che la proteina mutata Sod1 è espressa non solo nei motoneuroni, ma anche nei muscoli volontari dei soggetti malati. Tuttavia se questa espressione muscolare contribuisse all’induzione di qualche segno patologico della SLA rimaneva un punto irrisolto.
Molte erano quindi le domande ancora irrisolte. Infatti, come accennato precedentemente, l’ipotesi generale escludeva il muscolo scheletrico come bersaglio primario della malattia e stabiliva che l’atrofia e la debolezza muscolare, associati alla SLA, fossero dovute alla degenerazione dei neuroni motori. Quindi in assenza di alterazione neuronale, il muscolo sarebbe stato in grado di svolgere efficientemente le sue funzioni.
Una teoria questa che non ci convinceva; così abbiamo generato un nuovo modello sperimentale in cui il gene mutato della Sod1 è stato espresso selettivamente nei muscoli volontari. I risultati che abbiamo ottenuto hanno dimostrato infatti che nella Sla anche il muscolo fa la sua parte e che una alterazione muscolare determina un danno “retrogrado” sia a livello delle giunzioni neuromuscolari sia a livello del midollo spinale.
Un risultato questo che poteva essere raggiunto solo con dei modelli sperimentali animali. Nel nostro laboratorio utilizziamo ovviamente anche modelli sperimentali in vitro, come le colture 3D e laddove la sperimentazione animale si renda necessaria è importante sottolineare che vengono seguite regole molto precise che ne valutano non solo l'effettiva necessità, ma anche il modo con cui la ricerca verrà svolta, secondo i parametri di un codice etico molto rigido.
6) Quali sono le patologie neuromuscolari che potranno essere trattate (anche parzialmente) grazie ai progressi raggiunti con il vostro studio?
I risultati delle nostre ricerche hanno aggiunto un ulteriore tassello alla comprensione di patologie degenerative come la SLA, la distrofia muscolare e l'invecchiamento muscolare e nel contempo abbiamo suggerito una serie di "interventi" genetici e farmacologici per disegnare nuovi e più appropriati approcci terapeutici. Tanto resta tuttavia ancora da fare per passare dal bancone del laboratorio al letto del paziente.
7) Qual è e come funziona il meccanismo che ha permesso di preservare le giunzioni neuromuscolari dei topi trattati?
Il primo obiettivo è stato quello di individuare il meccanismo molecolare alla base dello smantellamento e abbiamo scoperto che questo dipende dall'attivazione di una proteina chinasi, conosciuta come PKC theta, la quale è normalmente coinvolta nel rifinimento delle giunzioni neuromuscolari nel corso dello sviluppo. Abbiamo dimostrato che questa proteina chinasi risultava "patologicamente" aumentata solo nei muscoli dei topilini che esprimevano la Sod1 mutata, ma non nei topolini normali (wild type).
La prova del nove che ha permesso di capire che fosse proprio questa aumentata attività della PKC-theta responsabile del danneggiamento delle giunzioni neuromuscolari è stata data da un esperimento di “silenziamento” farmacologico. Abbiamo infatti dimostrato che una inibizione farmacologica della proteina chinasi ha permesso di preservare le giunzioni neuromuscolari e di promuovere un mantenimento della massa e forza muscolare nei topolini trattati.
8) Ci spiega i concetti del “dying back” e del “saving back”, le rispettive differenze e come si applicano alle terapie per trattare le patologie neuromuscolari e la SLA?
Sulla base dei nostri studi pensiamo che la SLA potrebbe anche partire dai muscoli e, attraverso un meccanismo "retrogrado" definito appunto “dying-back”, portare ad una alterazione delle giunzioni neuromuscolari e del motoneurone.Pertanto, un approccio terapeutico che preservi il muscolo potrebbe portare anche a dei benefici a livello del motoneurone, attraverso appunto un meccanismo di "saving back" (salvataggio a ritroso, se così possiamo dire).
9) Come si modificheranno (se si modificheranno) le terapie per trattare la SLA e le altre patologie neuromuscolari? Nel prossimo futuro quali potrebbero essere gli scenari di cura per i malati di SLA e delle altre malattie neuromuscolari?
Sono profondamente convinto che per "aggredire" malattie come la SLA bisogna usare approcci combinati diretti verso le diverse componenti cellulari e tissutali coinvolte. Per esempio i nostri studi hanno contribuito enormemente a definire la SLA come una malattia multisistemica, che non riguarda soltanto i motoneuroni, ma che può coinvolgere direttamente anche altri tessuti (come la glia e il muscolo, appunto). È in questa nuova ottica, dunque, che bisogna cominciare a sviluppare approcci terapeutici combinati, che aggrediscano la malattia da più fronti.

